首页 / 产品和服务 / 产品类型 / 抗体产品 / 特色抗体推荐
Eukaryotic DNA is highly compacted and organized in nuclear space (1). To achieve this three-dimensional organization, DNA is wrapped around histone proteins in the form of chromatin (2, 3). Chromatin fibers with varying degrees of compaction and distinct types of chemical modification ensure proper gene expression and structural integrity of the genome (1). The integrity of chromatinized DNA is highly susceptible to both exogenous and endogenous sources of damage. Exogenous, or environmental damage can be produced by ultraviolet (UV) light from sunlight and ionizing radiation (IR, from, e.g., cosmic radiation and medical treatments employing X-rays or radiotherapy) as well as genotoxic carcinogens including topoisomerase inhibitors camptothecin (CPT) and etoposide, which inhibit topoisomerase I or II, respectively (4). The most frequent cell-intrinsic sources of genotoxic stress include free oxygen radicals as a result of increased cellular respiration, collapsed DNA replication forks as a consequence of abnormal cell division and variable joining of V, D and J gene segments or class switch recombination in vertebrate lymphocytes (5).
| ABclonal货号 | 产品名称 | 抗体来源 | 产品应用 | 应用物种 |
|---|
To counteract DNA damage, cells have evolved complex and elaborate mechanisms to repair different types of DNA lesions. Hundreds of repair genes have been identified, which mainly participate in five distinct but functionally interconnected pathways: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), non-homologous end joining (NHEJ) and homologous recombination (HR).
Single damaged bases resulting from chemically modifications can affect the ability of the bases to form hydrogen-bonds and lead to incorrect base-pairing. These mutations could be induced by IR, topoisomerase I poisons or some antimetabolites, and are largely repaired by BER (Figure 1A). The key components of the BER pathway are glycosylases, endonucleases, DNA polymerases and DNA ligases, with poly(ADP-ribose) polymerase 1(PARP1) and PARP2 facilitating the process (6). Damaged bases are first removed by glycosylases and endonucleases to form abasic sites and generate a 'nick' or a single-strand break, which can then be processed by either short-patch BER (where a single nucleotide is replaced) or long-patch BER (where 2-10 new nucleotides are synthesized) (7).
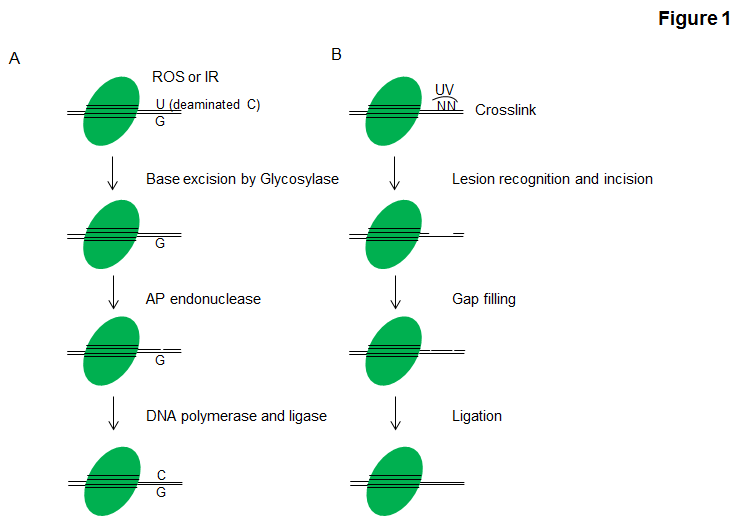
NER acts to remove helix-distorting adducts on DNA, for example, those caused by ultraviolet (UV) radiation and tobacco smoke. The xeroderma pigmentosum (XP) proteins and Cockayne syndrome associated proteins have crucial roles in this pathway (7). Eukaryotic NER can be divided into two subpathways: global genomic NER (GG-NER) and transcription coupled NER (TC-NER). For each subpathway, two distinct sets of proteins are involved in recognizing DNA damage (7). After that, the two subpathways converge for the steps of dual incision by Transcription factor II H (TFIIH), gap filling through DNA polymerases and ligation dependent on DNA ligase I and Flap endonuclease 1 or the Ligase-III-XRCC1 complex to seal the nicks to complete NER (Figure 1B).
MMR is a system for recognizing and repairing the daughter-strand specific replication errors that cause incorporation of the wrong nucleotide (a mismatch) and nucleotide insertions or deletions. The mispaired or fraudulent nucleotide is recognized by MSH2-MSH6 heterodimers, whereas deletions and insertions are recognized by MSH2-MSH3 heterodimers (6, 8). Besides specific MMR factors such as MLH1–MLH3 heterodimers required to process the downstream signaling, components of other DNA repair pathways including endonuclease 1, replication protein A (RPA), proliferating cell nuclear antigen (PCNA) and flap endonuclease 1 (FEN1) have also been implicated in excision and resynthesis past the lesion. Defective MMR increases mutation rates up to 1,000-fold, and results in microsatellite instability (MSI) thus cancer development (9).
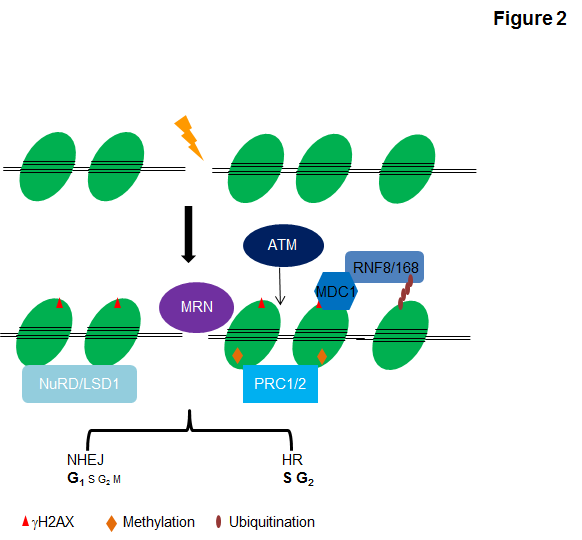
DSBs are among the most difficult lesions to repair and are profoundly cytotoxic, as the failure to repair DSBs can cause cell death, and their aberrant repair can lead to gross chromosomal changes. DSB repair generally occurs through one of two major pathways: nonhomologous end joining (NHEJ), which is by nature error-prone and ligates broken DNA with minimal end processing, or homologous recombination (HR), which restores the original DNA sequence using unperturbed template DNA, generally the sister chromatid (7, 9) (Figure 2). NHEJ is active in all phases of the cell cycle, predominating in G0 phase and G1 phase of the cell cycle, and it is considered to be responsible for the rapid repair of up to 85% of IR-induced DSBs. Although HR is generally limited to the S and G2 phase of the cell cycle and repairs only a minor proportion of DSBs, it may be the most crucial as it is high fidelity in dealing with stalled and collapsed replication forks, as well as single-ended DSBs, and also the processing of interstrand crosslinks (ICLs) in cooperation with the NER and Fanconi anaemia pathways (6). In HR, the broken DNA ends of a DSB are resected to allow invasion of the single strands into the sister chromatid, which functions as a spatially accessible template for accurate resynthesis of the damaged DNA (8).
Inappropriate repair of damaged DNA has deleterious effects not only on DNA lesion itself but other DNA transactions including DNA replication and gene transcription, and ultimately results in mutations and chromosomal aberrations (10). Thereby, the ability of cells to maintain genome integrity through orchestrating DNA damage response (DDR) temporally and spatially is vital for cellular homeostasis. The DDR is primarily mediated by proteins of the phosphatidylinositol 3-kinase-like protein kinase (PIKKs) family-ATM, ATR, and DNA-PK and by members of the PARP family-PARP1 and PARP2 (11). Through sensing/recognizing particular DNA lesions, these factors activate the DDR and set a choreographed response in motion to protect the cell and ameliorate the threat to the organism (4). Following or in concert with DDR activation, DNA repair is carried out by a plethora of enzymatic activities that chemically or physically modify repair proteins and chromatin structures in a reversible manner (Figure 2). These enzymes include but are not limited to nucleases, helicases, polymerases, topoisomerases, recombinases, ligases, glycosylases, methyltransferases, demethylases, ubiquitin ligases, deubiquitinases, kinases, and phosphatases (1, 12-16). In addition to these versatile enzymes, many DDR or repair processes involve scaffold proteins that lack inherent catalytic or DNA-binding functions, but function to recognize particular chemical modification via distinct reader domains or facilitate the association of functional subunits into multiprotein complexes. To timely respond to the damage stimuli and faithfully restore the breaks, these repair tools together with their modulators must be precisely regulated both temporally and spatially in the context of the surrounding chromatin, otherwise each in its own right can wreak havoc on the integrity of DNA as well as break proximal or distal chromatin configuration (17-19). Although a lot of work has been done to investigate cellular mechanisms that regulate the recruitment of DNA repair factors to sites of DNA damage and coordinate the choice of the pathways to employ for efficient DNA repair, the underlying mechanisms and implications of DNA repair in chromatin context are only beginning to be understood.
Defects in the maintenance of genome stability underlie a number of developmental disorders and human diseases including developmental defects, neurodegenerative diseases and cancers, which highlights the critical importance of an efficient DDR and repair program for cell and organism viability (11, 20-22). Classically, defects in the DDR and repair pathways have been taken advantage to exploit therapeutically in the treatment of cancer with radiation therapies or genotoxic chemotherapies. More recently, protein components that are amenable to modulation by small molecules among the DDR or DNA repair systems, have been identified as promising targets in cancer therapeutics.















