首页 / 产品和服务 / 产品类型 / 抗体产品 / 特色抗体推荐
Cell death is a critical and active process that maintains tissue homeostasis and eliminates potentially harmful cells. There are three major types of morphologically distinct cell death: apoptosis(type I cell death), autophagic cell death(type II), and necrosis(type III).
The characteristics of apoptosis include cell shrinkage, membrane blebbing, and condensation of the chromatin (pyknosis)[1] and apoptosis is often accompanied with the activation of caspase proteases[2].
Our initial understanding of the molecular mechanisms that control apoptosis came from early studies using the nematode Caenorhabditis elegans; these mechanisms were then extended to mammalian systems. A multitude of proteins and enzymes have now been identified that are involved in the initiation, amplification, or suppression of apoptosis[3].
Apoptosis maintains the healthy balance between cell survival and cell death through the contribution to elimination of unnecessary and unwanted cells[1, 4]. It is critical to animals especially long-lived mammals that must integrate multiple physiological as well as pathological death signals. Evidence indicates that accelerated cell death is evident in acute and chronic degenerative diseases, immunodeficiency and infertility, while insufficient apoptosis can manifest as cancer or autoimmunity. Under many stressful conditions like precancerous lesions, activation of the DNA damage checkpoint pathway can serve to remove potentially harmful DNA-damaged cells via apoptosis induction to block carcinogenesis[5, 6]. Thus, the evasion of apoptosis is a prominent hallmark of cancer [8]. Cancer cells are, in fact, harboring alterations that result in impaired apoptotic signaling, which facilitates tumor development and metastasis [6–8].
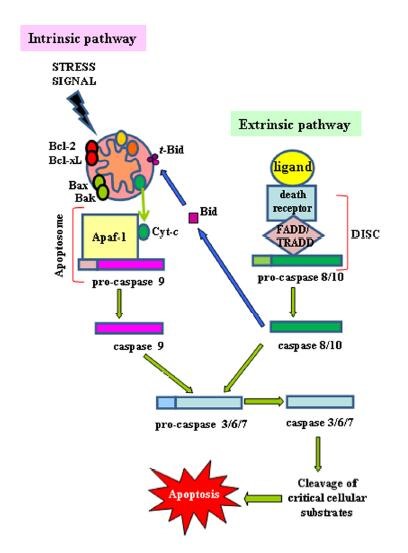
Two major signaling pathways trigger apoptotic cell death: the mitochondrial (the intrinsic) pathway and the death receptor (the extrinsic) pathway. The extrinsic pathway depends on binding of appropriate exogenous mediators to death receptors at the cell surface. In contrast, the intrinsic pathway responds to signals from within the cell, such as damages caused by radiation and various chemotherapeutic agents, to induce apoptotic signaling via the release of mitochondrial factors.
The intrinsic apoptotic pathway (mitochondriadependent) is mediated by intracellular signals that converge at the mitochondrial level in response to different stress conditions (i..e, irradiation, treatment with chemotherapeutic agents, etc.)[7]. Internal stimuli such as irreparable genetic damage, hypoxia, extremely high concentrations of cytosolic Ca2+ and severe oxidative stress are some triggers of the initiation of the intrinsic mitochondrial pathway[8]. Subsequent activation of pro-apoptotic BH3-only members of the Bcl-2 family (Bax, Bak) neutralizes the antiapoptotic proteins Bcl-2, Bcl-xL, and Mcl-1, leading to disruption of mitochondrial membrane outer membrane permeability (MOMP) so that proteins normally confined in the intermembrane space spread into the cytosol. These proteins include the so-called apoptogenic factors, such as cytochrome-c(CYCS), which plays a crucial role in activating the mitochondrial-dependent death in the cytosol[9]. Cytochrome-c binds to the cytosolic Apaf-1 (apoptosis protease activating factor-1) and triggers the formation of a complex named apoptosome, which recruits initiator pro-caspase-9 to its caspase recruitment domain (CARD), allowing autoactivation and then proteolysis. The process in turn activates downstream executor caspases-3, -6 and -7 for cleavage of cellular substrates leading to apoptotic cell death[9, 10].
Another mithochondrial component with a regulatory role in apoptosis is the apoptosis inducing factor (AIF)[11]. AIF has homology to bacterial oxidoreductases and is normally localized in the mitochondria, but translocates to the cytoplasm during apoptosis. Apoptosis induced by AIF cannot be inhibited by the caspase inhibitors, and can result in nuclear condensation and fragmentation independent of other pro-apoptotic factors. Interestingly, the redox-active enzymatic region of AIF is anti-apoptotic, giving the molecule a dual role in cell survival and cell death. The detailed pathways of caspase-independent apoptosis are poorly understood.
On the other hand, another group of molecules released by mitochondria including endonuclease G is also proapoptotic proteins but involved in the process at the later stages. These molecules are trans-located into the nucleus where they first cause an elementarily DNA fragmentation and chromatin condensation which is defined as “stage 1”, and an advanced condensation and DNA fragmentation by the help of caspase-3 at later stage is called as “stage 2”[12].
The endoplasmic reticulum (ER) is the central intracellular organelle in the secretory pathway. It is responsible for protein translocation, protein folding, and protein post-translational modifications that allow further transport of proteins to the Golgi apparatus and ultimately to vesicles for secretion or display on the plasma surface. Perturbations in ER function, a process named “ER-stress”, trigger the unfolded protein response (UPR), a tightly orchestrated collection of intracellular signal transduction reactions designed to restore protein homeostasis. The UPR is distinguished by the action of three signaling proteins named IRE1α (inositol-requiring protein-1α), PERK (protein kinase RNA (PKR)-like ER kinase), and ATF6 (activating transcription factor 6). Under physiological conditions, the luminal domains of PERK and ATF6 proteins are bound to the ER resident chaperone BiP (Binding immunoglobulin Protein), which keeps them inactive. When unfolded proteins accumulate in the ER, BiP is released from these complexes to assist with the folding of accumulated proteins. By comparison to the UPR modulators PERK and ATF6, which are modulated by association with BiP, IRE1α appears to become activated when unfolded proteins bind directly to it. Upon activation, PERK, IRE1α and ATF6 induce signal transduction events that alleviate the accumulation of misfolded proteins in the ER by increasing expression of ER chaperones, inhibiting protein entry into the ER by arresting mRNA translation, and stimulating retrograde transport of misfolded proteins from the ER into the cytosol for ubiquitination and destruction by a process named ERAD (ER-assisted degradation)[18].
In cases where ER stress cannot be reversed, cellular functions deteriorate, often leading to cell death. Accumulating evidence implicates ER stress-induced cellular dysfunction and cell death as major contributors to many diseases, making modulators of ER stress pathways potentially attractive targets for therapeutics discovery.
The ER lumen is the major storage of intracellular Ca2+ and Ca2+-binding chaperones mediate the proper folding of proteins in the lumen of the ER. It is well established that Ca2+ trafficking in and out of the ER regulates a diversity of cellular responses and signaling transduction pathways relevant to stress response, modulation of transcriptional processes, and development. For instance, acute release of Ca2+ from the ER can trigger a variety of signaling mechanisms that promote cell death mainly by Ca2+-mediated mitochondrial cell death. Conversely, pulses of Ca2+ delivered via IP3Rs at contact sites of ER and mitochondria promote oxidative phosphorylation, which sustains ATP levels and cell survival. Other proteins involved in ER Ca2+-mediated apoptosis are Bax and Bak[19].
The intrinsic and extrinsic apoptotic pathways are regulated by proteins such as p53 and NF-κB, or signaling pathways such as PI3K pathway and the ubiquitin proteosome system.
(1)p53
p53 as a transcription factor regulates downstream genes involved in cell cycle arrest, DNA repair, and apoptosis. Loss or mutation of p53 in many cancers leads to genomic instability, impaired cell cycle regulation, and inhibition of apoptosis. After DNA damage, p53 holds the cell at a checkpoint until the damage is repaired. If the damage is irreversible, apoptosis is triggered[20, 21].
(2)NF-κB
NF-κB functions as a nuclear transcription factor in regulating expression of a large number of genes involved in the regulation of apoptosis, viral replication, tumorigenesis, inflammation, and many autoimmune diseases[22] and growth factors, cytokines, lymphokines, radiation, pharmacologic agents, and stress could activate NF-κB[22]. Especially, NF-κB has been shown to have both anti- and proapoptotic functions that may be determined by the nature of the death stimulus rather than by the origin of the tissue.
(3)The ubiquitin/proteosome system
The ubiquitin/proteosome system is composed of a large proteinase complex that is responsible for the turnover of most intracellular proteins and consequently regulates cell growth and apoptosis[23]. Protein degradation is a highly coordinated process that involves recognition of the protein by attaching it to multiple ubiquitin molecules and then its digestion by the 26S proteosome. Many cell cycle regulators and transcription factors such as p53, cyclins and cyclin-dependent kinase inhibitors, and NF-κB are regulated by the ubiquitin/proteosome system[23, 24].
In addition, many of the Bcl-2 family members are substrates of the ubiquitin/proteosome[25]. The induction of apoptosis by proteosome inhibitors leads to an initial accumulation of proteins such as p53, p27, proapoptotic Bad or Bax, or activation of the stress kinase, which leads to the release of cytochrome-c and the activation of the intrinsic apoptosis pathway. This critical link between the apoptotic machinery and the ubiquitin/proteosome system has led to an increased interest in the design of inhibitory agents that target these pathways[25].
(4)PI3K
PI3K is a kinase that plays a central role in signaling pathways important to cell survival, proliferation, motility, and tissue neovascularization. PI3K is upregulated in many cancers[26]. Cell surface receptors induce the production of second messengers such as phosphatidylinositol 4,5-bisphosphate 3 and phosphatidylinositol 3,4,5- trisphosphate, which convey signals to the cytoplasm from the cell surface. Phosphatidylinositol 4,5-bisphosphate 3 signals activate the kinase 3-phosphoinositide-dependent protein kinase-1, which in turn activates the kinase Akt. Akt activation leads to phosphorylation of certain proteins that lead to cell survival[26]. Several proteins important in human cancers can be dysregulated in the PI3K pathway. These are also important because of Food and Drug Administration (FDA)-approved agents targeting them. For example, epidermal growth factor stimulation activates Akt via PI3K, as does HER-2/neu activation. Loss of phosphatase and tensin homolog tumor suppressor gene also augments the activity of this pathway[27].
(1)Caspase family members
Caspase family comprise conserved cysteine aspartic-specific proteases, and members of caspase family are considerably crucial in the regulation of apoptosis. There are 14 different caspases in mammals, and they are basically classified as the initiators including caspase-2, -8, -9, and -10; and the effectors including caspase-3, -6, -7, and -14; and also the cytokine activators including caspase-1, -4, -5, -11, -12, and - 13[28].
| Catalog | Name | Applications | Cross-Reactivity | Type |
|---|
Caspase-1, the first identified caspase, is interleukin-1b processing enzyme (ICE), and it is known as Ced-3 homologue. Caspase-1 is involved in cytokine activator group of caspase family since inflammatory cytokines, pro-IL-1b and pro-IL-18, are the main substrates for caspase-1. While caspase-1 is not essential for apoptotic signaling, it is essential in inflammation process.
The second identified caspase, caspase-2, containing CARD, plays important roles in DNA damage-, metabolic abnormality-, and ER stress-induced apoptosis. Caspase-2 is known as a substrate for both caspase-3 and caspase-8. Functional properties of caspase-2 have still not been clarified thoroughly.
Caspases-3, -6, and -7 are involved in the effector caspase group, and they act in a similar manner in the apoptotic process. Caspase-3 is activated via both extrinsic and intrinsic apoptotic pathways. Despite there are limited information about caspase-6 and -7 rather than caspase-3, it is known that while caspase-3 suppression results in the inhibition of apoptosis, suppression of caspase-6 and -7 do not significantly affect the apoptotic process. Furthermore, caspase-3 was reported to be crucial for PARP cleavage and DNA fragmentation which are hallmarks of apoptosis.
Functionally well-known member of caspase family, caspase-8, is crucial factor for TNF-induced extrinsic apoptotic pathway. Caspase-8, in turn, cleaves and activates caspase-3, -7, Bid, and also NF-kB.
Caspase-9, the initiator caspase, is an important factor for the generation of apoptosome complex in the mitochondrial pathway. Once cytochrome c is released from the mitochondria, it binds to Apaf1 which is the receptor for cytochrome-c in the cytoplasm. Cytochrome-c and Apaf-1 generate apoptosome, and then, pro-caspase-9 binds to Apaf-1. Afterward, pro-caspase-9 is activated via reciprocal cleavage, and by this way, apoptosome complex also become activated. Then, caspase-3 is cleaved and activated via caspase-9 found in the active apoptosome complex.
(2)Bcl-2 family members
Bcl-2 family members, which play important roles in regulating apoptotic signaling, are divided into three subfamilies including (i) pro-survival subfamily members (Bcl-2, BclXL, BclW, MCL1, and BFL1/A1), (ii) BH3-only subfamily members (Bad, Bim, Noxa, and Puma9), and (iii) pro-apoptotic mediator subfamily members (Bax and Bak). Basically, all of the members of Bcl-2 family share typical characteristic functions; (i) they dimerize with other members of Bcl-2 family, (ii) they contribute to the regulation of mitochondrial homeostasis by binding proteins, and (iii) they contribute to outer mitochondrial membrane pore formation[29].
In several types of human malignancies, the balance between the expression levels of Bcl-2 family genes is broken down, and the equilibrium changes to the pro-survival subfamily member direction. In this case, cancer cells can escape from apoptotic signals and therefore develop resistance against therapeutic agents. Additionally, Bcl-2 family members are also considered in cancer therapy due to their therapeutic potentials. In the clinical trials, the BH3-only mimetic agents targeting Bcl-2 are being investigated in order to find alternative potent therapeutic approaches in several types of malignancies[30].
The selective modulation of both apoptotic pathways has proven to be a challenge in cancer drug development. Unlike most oncogenes that work by promoting proliferation, Bcl-2 functions by preventing programmed cell death. These proteins therefore provide therapeutic targets where their inhibition can lead to the induction of apoptosis[31].
(1)Targeting the intrinsic pathway
Bcl-2 inhibitors
The anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-XL, Bcl-w,and MCL1) are up-regulated in various cancers including B-cell lymphoma and other haematological malignancies, and small cell lung cancer(SCLC). The drugs targeting the Bcl-2 proteins are further studied deeply in tumor therapy[32].
Targeting the BH3 domain of Bcl-2
One major strategy against Bcl-2 has been the development of small molecules that target BH3. These small molecules mimic the action of BH3 proteins and interact with anti-apoptotic proteins by binding their BH-3 binding groove. They include gossypol, ABT-737,ABT263 (navitoclax), ABT-199 and obatoclax.
(2)Targeting the extrinsic pathway
Pro-apoptotic receptor agonists (PARAs)
Tumor necrosis factor-related apoptosis-inducing ligand(TRAIL/Apo2L) is a member of the tumor necrosis factor ligand superfamily[33, 34]. TRAIL induces apoptosis in malignant cell lines via its two death receptors, TRAIL-R1 and TRAIL-R2. TRAIL seems to have minimal activity against most normal cell types but TRAIL-R1 and TRAIL-R2 are expressed at high levels in a variety of solid tumors[35, 36]. Pre-clinical data demonstrated that agonistic antibodies against TRAIL receptors induced apoptosisin a variety of tumor types while sparing the normal tissue, making this approach a relevant strategy in targeting cancer [36]. Pro-apoptotic receptor agonists (PARAs), which include antibodies against TRAIL-Rs and recombinant human (rh) TRAIL antibody, also showed synergistic effects when combined with chemotherapy and radiotherapy in several pre-clinical models[37]. This led to several clinical studies which are on-going in single agent and combination settings.
Pan recombinant human TRAIL (rh-TRAIL)antibodies
Dulanermin is a recombinant human TRAIL antibody targeting both TRAIL-R1 and TRAIL-R2. Pre-clinical studies demonstrated selective induction of apoptosis in cancer cells leading to caspase activation and subsequent cell death[36]. Several pre-clinical studies showed activity of this compound as a single agent and in combination with chemotherapy in solid and haematological malignancies[38].
TRAIL-R1 agonistic monoclonal antibodies
Mapatumumab is a fully humanimmunoglobulin G1 lambda (IgG1 λ) monoclonal agonistic antibody(mAb) targeting TRAIL-R1. Pre-clinical datademonstrated that mapatumumab induced tumor regressionin mice bearing established TRAIL-R1-expressing human tumor xenografts. As a single agent, mapatumumab was ableto enhance the anti-tumor activity of cytotoxic agents in several cell lines, including those resistant to chemotherapy[39]. Currently, Mapatumumab is under evaluation in several combination phase II studies, including with cisplatin and radiotherapy in patients with cervical cancer and with bortezomib in patients with refractory myeloma.
TRAIL-R2 agonistic monoclonal antibodies
Lexatumumab is a high-affinity recombinant fully human IgG1λ mAb that binds to andactivates TRAIL-R2. Preclinical data indicated activity inrenal, hematological, breast, ovarian and CRC cells lines and xenografts[40].
Conatumumab (AMG 655) is a monoclonal antibody specific for TRAIL-R2 that has been shown to activate the caspases in vivo and in vitro in human cancer models, both as a single agent and in combination with chemotherapy[41]. The antibody has been evaluated in two phase I trials[42].
Other agonistic TRAIL-R2 antibodies
Drozitumab, tigatuzumab and LBY135. These agonist TRAIL-R2 antibodies have been evaluated in the phaseI/II setting. Drozitumab has been investigated into chondrosarcoma, CRC and granulosa cell tumor[43]. Tigatuzumab[44] and LBY135[45] has also been in the clinical research for the therapy.
(3)Targeting the intrinsic pathway
IAPs are abnormally regulated and over-expressed in avariety of human malignancies and are also known to promotethe invasion and metastases of cancer cells[46, 47]. There are currently two approaches to targeting IAPs, which include(i), targeting IAPs by mimicking Smac, an inhibitor of IAPs,and (ii), anti-sense mediated interference of XIAP mRNA and protein expression.
With the important roles of apoptosis in regualting the balance of cell survival and cell death, especially associated with the occurrence and development of tumor, the drugs targeting the apoptosis pathway have brought us into a new perspective into the tumor therapy. However, it is not enough and clear for us to understand the specific mechanism of apoptosis pathway in the occurrence and development of tumor, such as some key regulatory factors, so further more studies would be done for finding the better targeting drugs in the treatment of cancers.


![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_H1110_WB_01.jpg?t=1775203334 "alt=")
![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_P457_KO-WB_01.jpg?t=1775203334 "alt=")
![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_P457_IF_01.jpg?t=1775203334 "alt=")
![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_P457_IF_02.jpg?t=1775203334 "alt=")
![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_P457_IF_03.jpg?t=1775203334 "alt=")
![[KO Validated] AIF Rabbit pAb](https://pic.abclonal.com.cn/A2568_P457_IF_04.jpg?t=1775203334 "alt=")



