产品信息
产品描述
ABScript II cDNA First Strand Synthesis Kit 包含 ABScript II Enzyme Mix 和 ABScript II Reaction Mix 两种经过优化的Mix。其中 ABScript II Enzyme Mix 包含了 ABScript II 逆转录酶和 RNase 抑制剂,而 ABScript II Reaction Mix 含有优化的反应 Buffer。 ABScript II 逆转录酶是一种重组 M-MuLV 逆转录酶,拥有较低的 RNase H 活性同时兼具增强的热稳定性。与野生型 M-MuLV 相比,重组 M-MuLV 逆转录酶可以在更高的温度下合成第一链 cDNA。该酶在高达 48℃时仍具有活性,特异性 cDNA 产量更高。
该试剂盒提供两种逆转录引物。Oligo-dT 引物[d(T)23VN]迫使引物退火至 Poly(A)尾的起始处。优化的 Random Primer Mix特异性较低,因此所有 RNA,包括 mRNA、rRNA、tRNA 均可作为逆转录模板。本试剂盒可合成长达 10 kb 的 cDNA。
产品特点
- 较低的RNase H活性利于长片段cDNA合成,可以合成长达10 kb的cDNA;
- 酶最适温度是42℃并且在48℃时仍具有较高的活性;
- 比普通M-MLV反转录酶(37℃)特异性cDNA产量更高;
- 适合新冠病毒检测;
- 配套多种RT引物,mRNA和ncRNA均能作为逆转录模板;
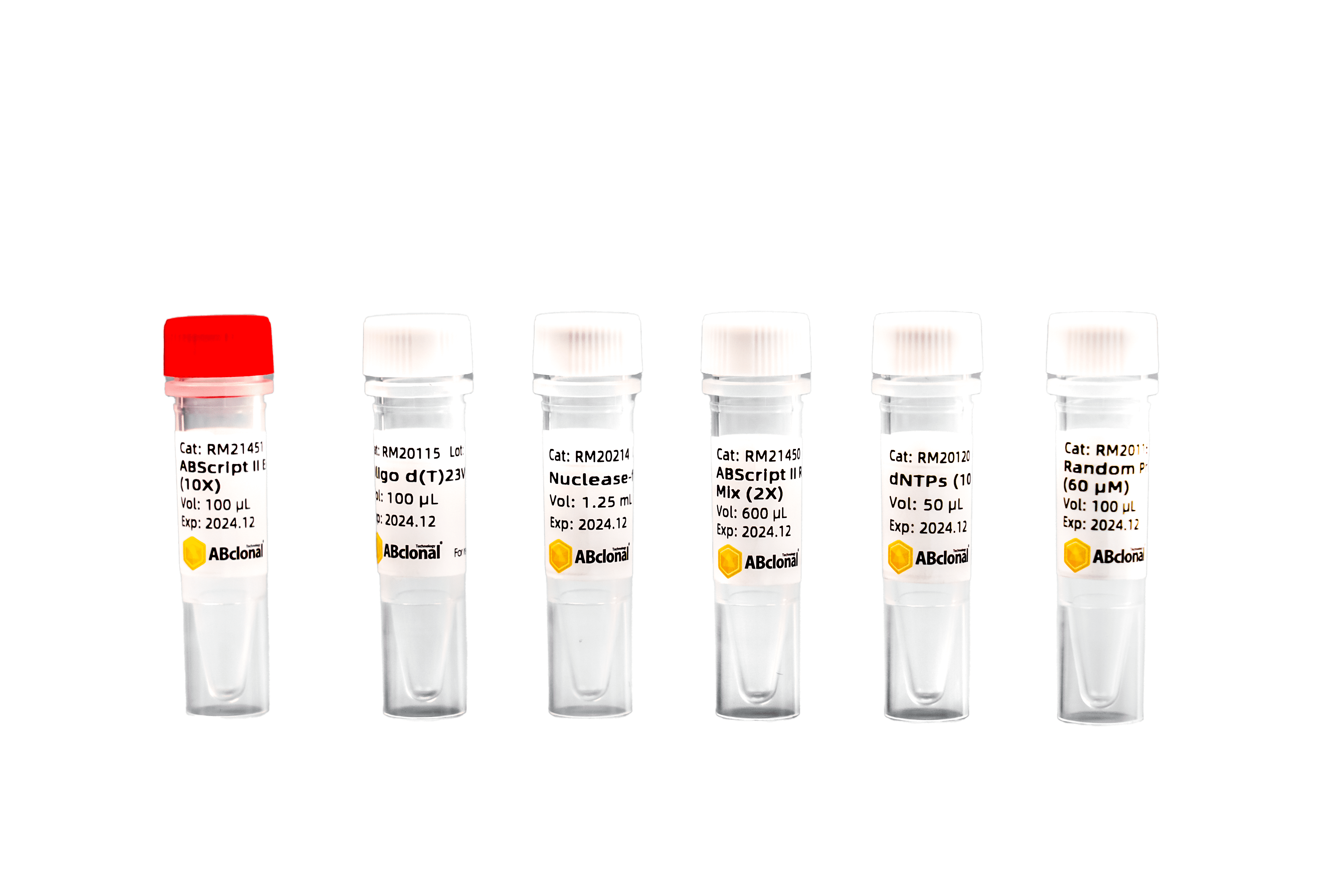
产品组分
| 试剂盒组成 | 50 RXN
(20 μL / RXN) | 100 RXN
(20 μL / RXN) |
| ABScript II Enzyme Mix (10X) | 100 µL | 200 µL |
| ABScript II Reaction Mix (2X) | 600 µL | 1.25 mL |
| Oligo d(T)23VN (50 μM) | 100 µL | 200 µL |
| Random Primer Mix (60 μM) | 100 µL | 200 µL |
| dNTPs (10 mM each) | 50 µL | 100 µL |
| Nuclease-free H2O | 1.25 mL | 1.25 mL |
FAQs
Publishing research using RK20400? Please let us know so that we can cite the reference in this datasheet.
说明书
说明书下载